最近看到好多有关计算有效质量的帖子。在论坛内搜了下,发现很多都侧重于软件具体操作,而对单位准换等细节问题讲述的较少。因此鄙人结合自己对有效质量的理解,阐述下自己想到的一种计算有效质量的方法。不足之处,还望多多指正。
有效质量的表达式,一般固体物理教材上都有:
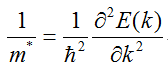
在带入物理量进行计算时,涉及单位制问题。一般写输入文件时,长度单位为埃;而程序输出的能带结构中,能量单位为eV(abinit是个例外,默认输出能量单位为hartree)。这两个单位实际上是国际单位制下的单位,在此单位制下计算不仅要查找约化普朗克常数h_bar的值,最后算得有效质量单位为千克,还得换算成电子质量倍数,非常麻烦。而改用原子单位制就可省去这些步骤。因此算有效质量第一步就是将所有物理量转换成原子单位制。
原子单位制有两种,一种为Hartree原子单位制,另一种为Rydberg单位制。这两种单位制的区别在于,Hartree单位制下基本物理量简单,电子电荷和质量都为1;而Rydberg单位制下薛定谔方程简单,系数为1。由于Rydberg单位制下电子质量定义为1/2,这不太符合一般认知规律(就像某些编程语言中从0开始的数组下标一样,扯远了),因此一般转换成Hartree单位制。Hartree单位制下,一长度单位等于1bohr等于0.52917721092埃,一能量单位等于27.21138506eV,约化普朗克常数值为1。这样有效质量表达式中的约化普朗克常数就没了。
第二步,是根据原胞基矢和正倒格子基矢间对应关系,算出倒格子基矢。
以单层二硫化钼为例,其为六方晶系,原胞基矢长度为3.20埃,c轴设定为10埃。在以埃为单位长度的笛卡尔坐标下,三个正格子基矢的坐标为:
a1 = 1.600000000 2.771281292 0.000000000
a2 = -1.600000000 2.771281292 0.000000000
a3 = 0.000000000 0.000000000 10.000000000
实际上正格子基矢在笛卡尔坐标系中有无数种摆法,这里选取了其中一种,让a1,a2对称分布在y轴两侧。
将坐标转换成原子单位制:
a1 = 3.023561799 5.236962656 0.000000000
a2 = -3.023561799 5.236962656 0.000000000
a3 = 0.000000000 0.000000000 18.897261246
再根据公式:
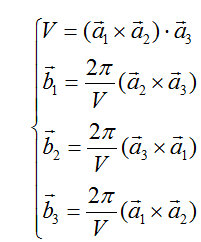
算得倒格子基矢坐标为:
b1 = 1.039037024 0.599888305 -0.000000000
b2 = -1.039037024 0.599888305 0.000000000
b3 = 0.000000000 -0.000000000 0.332491848
对于六角晶系,如果正格子基矢夹角为60度,倒格子则为120度。
取一条K路径:
G: (0, 0, 0) # Gamma
M: (1/2, 0, 0)
K: (2/3, 1/3, 0)
以上各K点坐标为分数坐标,换算成笛卡尔坐标:
G: ( 0, 0, 0 )
M: ( 0.5195185, 0.2999442, 0.0 )
K: ( 0.3463457, 0.5998883, 0.0 )
各K点间距:
|GM| = 0.5998883 bohr^-1, |MK| = 0.3463456 bohr^-1, |KG| = 0.6926914 bohr^-1。
在计算能带时各高对称点间均匀撒点。按图中方法确定从出发点到当前点所走过的路径长度,最为自变量x:
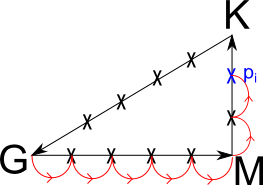
将能量转换成以Hartree为单位后,作为y。
定位价带顶和导带底,对那一段进行多项式拟合,得到y与x的表达式(实际上是E与k的表达式)。
将y对x求二阶导数,计算在价带顶和导带底的导数值(如果用二次函数拟合,二阶导数应该为常数),再取其导数,即有效质量。
此有效质量单位是电子质量。例如结果是0.5,则表示有效质量为电子质量0.5倍。
最后附带一个由正格子基矢算倒格子基矢的python脚本
http://emuch.net/bbs/viewthread.php?tid=7149817&fpage=1
|









 QQ好友和群
QQ好友和群 QQ空间
QQ空间 腾讯微博
腾讯微博 腾讯朋友
腾讯朋友 收藏
收藏 淘帖
淘帖 顶
顶 踩
踩


